General Usage¶
The purpose of RiboPipe is to automate the alignment, quality control, and initial analysis of ribosome profiling and other short, single-end read data. It is intended that
input data is a directory of .fastq formatted files. However, when using the intermediate submodules, such as align or quality, input will vary and is explicated
in the --help menu for each submodule.
RiboPipe was created with the novice bioinformatician in mind, and the documentation written assuming no background in using Command Line or programming. As such, instructions have been created with as much detail as possible. Additionally, `walkthrough videos <>`_ are available. In cases where RiboPipe is being used on a cloud computing cluster, or Linux machine with Singularity installed, a singularity container image can be used to avoid the installation process. Additionally, alignment references have been created for a variety of model organisms using current builds to help users avoid this step. Details on how to incorporate a newly created reference not already provided within the RiboPipe infrastructure can be found here.
RiboPipe can be run essentially from beginning to end as a pipeline, or as individual submodules. We will describe each option in detail below.
Important File Naming Conventions¶
In order for many of the RiboPipe functions to perform properly and for the output to be reliable after alignment (except for generation of a raw counts table), file naming conventions must be followed.
- Download your raw sequence data and place in a folder – this folder should contain all the sequence data and nothing else.
- Make sure files follow a pattern naming scheme. For example, if you had 3 genetic backgrounds of ribosome profiling data, the naming scheme would go as follows:
ExperimentName_BackgroundA_FP.fastq(.qz)
ExperimentName_BackgroundA_RNA.fastq(.qz)
ExperimentName_BackgroundB_FP.fastq(.qz)
ExperimentName_BackgroundB_RNA.fastq(.qz)
ExperimentName_BackgroundC_FP.fastq(.qz)
ExperimentName_BackgroundC_RNA.fastq(.qz)
- If the sample names are replicates, their sample number needs to be indicated.
- If you want the final count table to be in a particular order and the samples ordered that way are not alphabetically, append a letter in front of the sample name to force this ordering.
ExperimentName_a_WT_FP.fastq(.qz)
ExperimentName_a_WT_RNA.fastq(.qz)
ExperimentName_b_exType_FP.fastq(.qz)
ExperimentName_b_exType_RNA.fastq(.qz)
- If you have replicates:
ExperimentName_a_WT_1_FP.fastq(.qz)
ExperimentName_a_WT_1_RNA.fastq(.qz)
ExperimentName_a_WT_2_FP.fastq(.qz)
ExperimentName_a_WT_2_RNA.fastq(.qz)
ExperimentName_b_exType_1_FP.fastq(.qz)
ExperimentName_b_exType_1_RNA.fastq(.qz)
ExperimentName_b_exType_2_FP.fastq(.qz)
ExperimentName_b_exType_2_RNA.fastq(.qz)
- If you are just running RNAseq files through the pipeline, you only need the RNA samples in your input directory and specify the rnaseq module:
ExperimentName_a_WT_1_RNA.fastq(.qz)
ExperimentName_a_WT_2_RNA.fastq(.qz)
ExperimentName_b_exType_1_RNA.fastq(.qz)
ExperimentName_b_exType_2_RNA.fastq(.qz)
ribopipe rnaseq -i input_directory ...
riboseq module¶
Pipeline for handling raw ribosome profiling sequence data. Runs quality and adaptor trimming, alignment, quality control, and formatting on a directory of raw ribosome profiling sequence data.
The riboseq module can be run as follows:
$ ribopipe riboseq -i $DATA_PATH/raw_ingolia/ -o $DATA_PATH/out_ingolia/ -r yeast -e ingolia_2015 \
-s a_WT_DED1_1_15deg b_WT_DED1_2_15deg c_ded1_cs_1_15deg d_ded1_cs_2_15deg \
e_WT_DED1_1_37deg f_WT_DED1_2_37deg g_ded1_ts_1_37deg h_ded_ts_2_37deg \
i_WT_TIF1_1_30deg j_WT_TIF1_2_30deg k_tif1_ts_1_30deg l_tif1_ts_2_30deg \
m_WT_TIF1_1_37deg n_WT_TIF1_2_37deg o_tif1_ts_1_37deg p_tif1_ts_2_37deg \
-p HISAT2 -a CTGTAGGCACCATCAAT --platform ILLUMINA --count_cutoff 32
An explanation of the riboseq submodule arguments can be found in the following tables:
| Argument | Description |
|---|---|
-i INPUT, --input INPUT |
Specify full PATH to input directory |
-o OUTPUT, --output OUTPUT |
Specify full PATH to output directory |
-r <yeast>, <human>, <mouse>, --reference <yeast>, <human>, <mouse> |
Specify model organism used for experiments. Pipeline will align a current, curated reference |
-e <string>, --experiment <string> |
Provide experiment name to prepend to output files |
-s <string> [<string> ...], --samples <string> [<string> ...] |
Space delimited list of samples in order. If replicates are included, indicate number in sample name to delineate. |
-a <string> [<string> ...], --adaptor <string> [<string> ...] |
Sequence of 3’ linker (only supports one 3’ linker currently) (default: AACTGTAGGCACCATCAAT). If no adaptor was used, specify None’ |
| Argument | Description |
|---|---|
-m <int>, --max_processors <int> |
Number of max processors pipeline can use for multiprocessing tasks (default: No limit) |
-f, --footprints_only |
Select this option if ONLY providing raw footprint sequence data |
--min_overlap <integer> |
Minimum number of bases that must match on a side to combine sequences for rrna_prober |
-p <HISAT2>, <STAR>, --program <HISAT2>, <STAR> |
Alignment software to be used to align reads to reference (default: STAR) |
--read_length_min <int> |
Minimum read length threshold to keep for reads (default: 11) |
--read_length_max <int> |
Maximum read length threshold to keep for reads (default: 50) |
--read_quality <int> |
PHRED read quality threshold (default: 28) |
--platform <SANGER>, <ILLUMINA> |
Sequencing platform used (default: ILLUMINA) |
--full_genome |
Add this option to map reads to full genome. If not given, will not count any read mapping to the first 45 nt of transcripts for ribosome profiling |
--count_cutoff <int> |
Minimum counts threshold. Will remove any row in the final count tables if any sample does not meet this cutoff threshold |
Additional information:¶
-s, --samples: For ribosome profiling, do not differentiate between footprint and RNA samples.
--full_genome: It is recommended to NOT include this option as ribosome profiling data for this region is often unreliable.
Follow up with the diffex submodule for differential expression analysis.
rnaseq module¶
Similar to the riboseq module, but tuned for small RNAseq (i.e. anything single-end under 100 bps). It is expected that in the future, RiboPipe will be able to perform
automated paired-end and genome sequencing assembly and alignment.
The rnaseq module can be run as follows:
$ ribopipe rnaseq -i $DATA_PATH/raw_dang_berger/ -o $DATA_PATH/out_dang_berger/ -r yeast -e dang_berger_2014 \
-s WT_NR_A WT_NR_B WT_CR_A WT_CR_B \
-p HISAT2 -a TGGAATTCTCGGGTGCCAAGG --platform ILLUMINA --replicates
An explanation of the riboseq submodule commands can be found in the following tables:
| Argument | Description |
|---|---|
-i INPUT, --input INPUT |
Specify full PATH to input directory |
-o OUTPUT, --output OUTPUT |
Specify full PATH to output directory |
-r <yeast>, <human>, <mouse>, --reference <yeast>, <human>, <mouse> |
Specify model organism used for experiments. Pipeline will align a current, curated reference |
-e <string>, --experiment <string> |
Provide experiment name to prepend to output files |
-s <string> [<string> ...], --samples <string> [<string> ...] |
Space delimited list of samples in order. If replicates are included, indicate number in sample name to delineate. |
-a <string> [<string> ...], --adaptor <string> [<string> ...] |
Sequence of 3’ linker (only supports one 3’ linker currently) (default: AACTGTAGGCACCATCAAT). If no adaptor was used, specify None’ |
| Argument | Description |
|---|---|
-m <int>, --max_processors <int> |
Number of max processors pipeline can use for multiprocessing tasks (default: No limit) |
--replicates |
Select this option if samples are replicates (do not use if 3+ replicates). |
-p <HISAT2>, <STAR>, --program <HISAT2>, <STAR> |
Alignment software to be used to align reads to reference (default: STAR) |
--read_length_min <int> |
Minimum read length threshold to keep for reads (default: 11) |
--read_length_max <int> |
Maximum read length threshold to keep for reads (default: 50) |
--read_quality <int> |
PHRED read quality threshold (default: 28) |
--platform <SANGER>, <ILLUMINA> |
Sequencing platform used (default: ILLUMINA) |
--full_genome |
Add this option to map reads to full genome. If not given, will only map reads to transcripts. |
--count_cutoff <int> |
Minimum counts threshold. Will remove any row in the final count tables if any sample does not meet this cutoff threshold |
Additional information:¶
--replicates: Make sure when passing the argument to list samples these are sorted so replicates are next to each other in this list
Follow up with the diffex submodule for differential expression analysis.
trim module¶
Perform only trimming and quality control steps, outputting general read information. Will output trimmed reads ready for alignment.
The trim module can be run as follows:
$ ribopipe trim -i $DATA_PATH/input_files/ -o $DATA_PATH/output_files/ \
-a TGGAATTCTCGGGTGCCAAGG --platform ILLUMINA
An explanation of the trim submodule commands can be found in the following tables:
| Argument | Description |
|---|---|
-i INPUT, --input INPUT |
Specify full PATH to input directory |
-o OUTPUT, --output OUTPUT |
Specify full PATH to output directory |
-a <string> [<string> ...], --adaptor <string> [<string> ...] |
Sequence of 3’ linker (only supports one 3’ linker currently) (default: AACTGTAGGCACCATCAAT). If no adaptor was used, specify None’ |
| Argument | Description |
|---|---|
-m <int>, --max_processors <int> |
Number of max processors pipeline can use for multiprocessing tasks (default: No limit) |
--read_length_min <int> |
Minimum read length threshold to keep for reads (default: 11) |
--read_length_max <int> |
Maximum read length threshold to keep for reads (default: 50) |
--read_quality <int> |
PHRED read quality threshold (default: 28) |
--platform <SANGER>, <ILLUMINA> |
Sequencing platform used (default: ILLUMINA) |
align module¶
Perform only alignment of input files – assumes files have already been trimmed if required. Will output alignment files and genome browser compatible files.
The align module can be run as follows:
$ ribopipe align -i $DATA_PATH/input_files/ -o $DATA_PATH/output_files/ -r yeast -e align_only_test \
-s sample1_FP sample1_RNA sample2_FP sample2_RNA
-a TGGAATTCTCGGGTGCCAAGG --platform ILLUMINA
An explanation of the align submodule commands can be found in the following tables:
| Argument | Description |
|---|---|
-i INPUT, --input INPUT |
Specify full PATH to input directory |
-o OUTPUT, --output OUTPUT |
Specify full PATH to output directory |
-r <yeast>, <human>, <mouse>, --reference <yeast>, <human>, <mouse> |
Specify model organism used for experiments. Pipeline will align a current, curated reference |
-e <string>, --experiment <string> |
Provide experiment name to prepend to output files |
-s <string> [<string> ...], --samples <string> [<string> ...] |
Space delimited list of samples in order. If replicates are included, indicate number in sample name to delineate. |
t <riboseq>, <rnaseq>, --type <riboseq>, <rnaseq> |
Sequencing type – riboseq for ribosome profiling or rnaseq for single-end short read sequence data. |
| Argument | Description |
|---|---|
-m <int>, --max_processors <int> |
Number of max processors pipeline can use for multiprocessing tasks (default: No limit) |
-p <HISAT2>, <STAR>, --program <HISAT2>, <STAR> |
Alignment software to be used to align reads to reference (default: STAR) |
--full_genome |
Add this argument if you wish to align to the full genome. See notes in riboseq and rnaseq sections above for more information. |
--count_cutoff <int> |
Minimum counts threshold. Will remove any row in the final count tables if any sample does not meet this cutoff threshold |
quality module¶
Takes a table of raw counts generates paired scatter plots.
The quality module can be run as follows:
$ ribopipe quality -i $DATA_PATH/raw_counts.csv -o $DATA_PATH/output_location/ \
-t riboseq
An explanation of the quality submodule commands can be found in the following tables:
| Argument | Description |
|---|---|
-i INPUT, --input INPUT |
Input table (must be .csv file) of raw counts |
-o OUTPUT, --output OUTPUT |
Specify full PATH to output directory |
-t <riboseq>, <rnaseq>, --type <riboseq>, <rnaseq> |
Sequencing type – riboseq for ribosome profiling or rnaseq for single-end short read sequence data. |
Additional information:¶
Table must have samples set as columns and must be ordered as sample1_FP, sample1_RNA, sample2_FP, sample2_RNA, etc. or as sample1_rep1_RNA, sample1_rep2_RNA, sample2_rep1_RNA, sample2_rep2_RNA.
rrna_prober module¶
As the bulk of total cellular RNA consists mostly of ribosomal RNA that is often not of interest and crowds out measurement of other RNAs, an rRNA depletion step is commonly performed before
creating a cDNA sequencing library. Several commercial kits are available that can effectively deplete samples of full length rRNAs. However, inherent in ribosome profiling protocols, samples are
treated with an RNase, which degrades full length rRNAs, making them more difficult to deplete before creating the cDNA library. One way to better deplete these rRNA fragments in ribosome profiling
samples is to create biotinylated RNA probes for overrepresented rRNA fragments to extract these fragments (see this paper for a recent discussion).
rrna_prober automates the identification of overrepresented species and outputs a table with these sequences ordered by abundance for ease of probe generation. This table is output in stdout
in this module, or as a txt file in the highlights/ output folder when running the full riboseq submodule.
The rrna_prober module can be run as follows:
$ ribopipe rrna_prober -i $DATA_PATH/sample1.zip $DATA_PATH/sample2.zip ... \
-o $DATA_PATH/output_location/
An explanation of the rrna_prober submodule commands can be found in the following tables:
| Argument | Description |
|---|---|
-i <string> [<string> ...], --input <string> [<string> ...] |
Space delimited list of zipped files (include full paths to these files) |
-o <string>, --output <string> |
Output file name to write output to |
gene_dictionary module¶
diffex module¶
truncate module¶
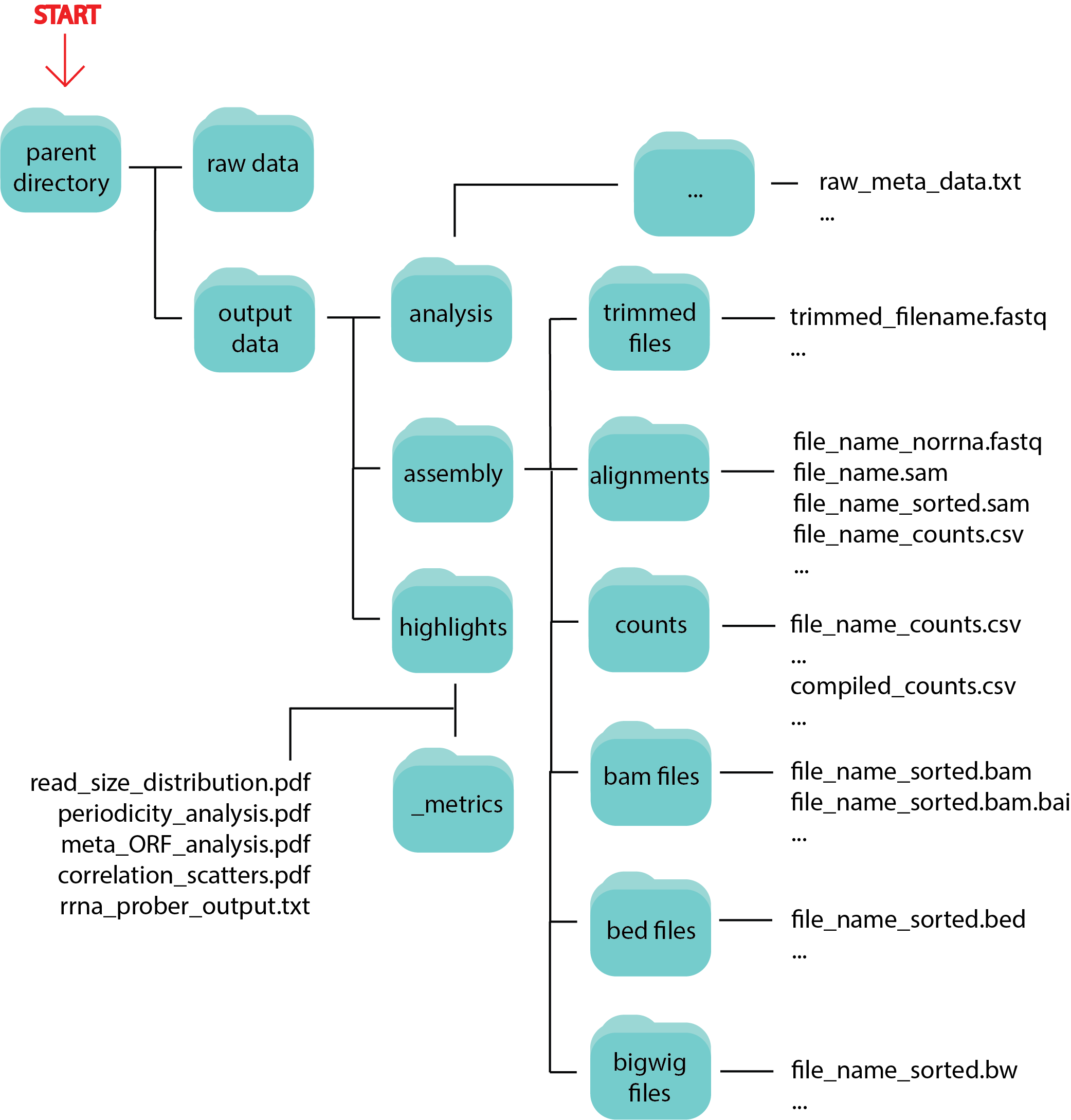
Data Output¶
Running riboseq or rnaseq will output all intermediate and final data files in a tree structure as seen below: